Research in the group focuses on the study of soft-matter systems (proteins, polymers, and a number of biological materials) using the principles of Statistical Mechanics and Polymer Physics We use a combination of theory and molecular Simulations (Monte Carlo and Molecular Dynamics) to draw insights into the interactions that take place at the nano-scale.
By adopting a multi-scale modeling approach, we are able to understand both the qualitative ( by using coarse-grained simulations) and quantitative (Atomistic Molecular Dynamics) understanding of the systems.
1) Building stimuli-responsive nanoparticle self-assemblies in solutions
Nanoparticle self-assembly in solution has gained immense interest due to the enhanced optical, chemical, magnetic and, electrical properties which manifest at the macroscale. The enhancement of the physical properties is a direct consequence of the structural morphology of the nanoparticle in solutions. We use a combination of molecular dynamics simulations and mean-field theory to understand and tune the self-assembly of nanoparticles grafted with polyelectrolyte chains. Our study shows that by tuning the graft density, the chain length, and the charge density of the grafts, we could build and control a variety of self-assembled structures ranging from rings, dimers, strings, coil-like aggregates, disordered to ordered aggregates.

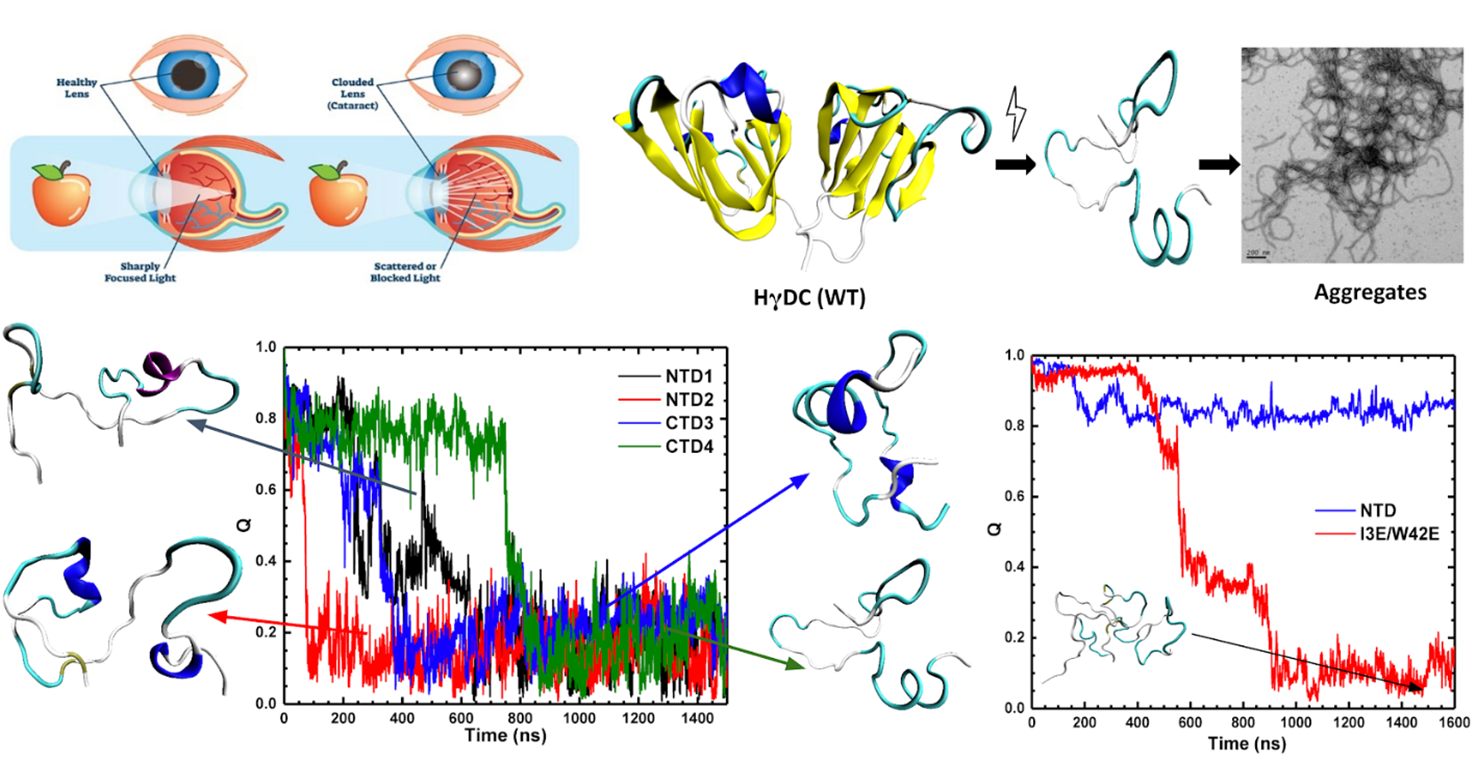
2) Understanding the aggregation pathways of Human Crystallin Protein
Age onset cataract is a common protein aggregation disease affecting 25 million people worldwide. The Human eye lens is made of $\alpha$, $\beta$, and $\gamma$ crystallin proteins. These proteins are responsible for the transparency and refractive index of our eye lens. The partial unfolding/aggregation of these proteins leads to opacification of the lens commonly known as Cataract. Human $\gamma$D proteins are the most abundant $\gamma$ crystallin proteins in the lens. The current study focuses on understanding the unfolding behavior of the $\gamma$D crystallin protein and the effect of mutations that lead to aggregation. We are using all atomistic Molecular Dynamics simulations with a combination of Markov Models to understand the unfolding behavior on a long time scale.
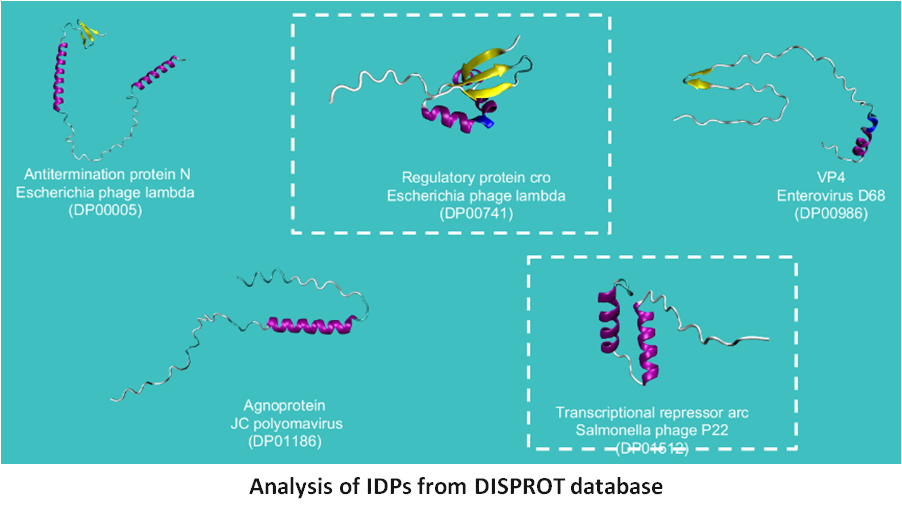
3) Modeling and prediction of the structure of intrinsically disordered peptides (IDPs)
Charge density and distribution play a vital role in the structure and functions of proteins. IDPs are proteins/regions of proteins that lack a well-defined secondary structure like helices or sheets. Further, the structure of IDPs is very dynamic in nature and hence the name "Dancing Proteins has been associated with them. Their sequences are deficient in hydrophobic groups and enriched in polar and charged residues and are mostly ampholytes (about 75% of found to date). The current project uses a combination of theory, molecular simulations, and bioinformatics to understand the predict the structure and function of IDPs using charge distribution as a key metric.
Modeling and Analysis of Supercapacitor using Molecular Dynamics Simulation and Graph Theory
Supercapacitors are the energy storage devices that bridge the gap between high power density (capacitor) and high energy density (battery) systems. The current project involves the design of a novel engineered porous electrode with a high specific surface area using the tools of molecular dynamics simulations aided by Graph Theory

Dabbling with Financial Modelling